deconvolve
This command performs idnetification/deconvolution of hit peptides and their activities from pooled ELISpot readouts.
usage: ace deconvolve [-h]
--readout-file-type READOUT_FILE_TYPE
--readout-files READOUT_FILES
--assignment-excel-file ASSIGNMENT_EXCEL_FILE
--min-pool-spot-count MIN_POOL_SPOT_COUNT
--output-excel-file OUTPUT_EXCEL_FILE
[--method {em,lasso,cem,empirical}]
[--background-spot-count BACKGROUND_SPOT_COUNT]
[--verbose VERBOSE]| Required Parameter | Description |
|---|---|
--readout-file-type |
ELISpot readout file type (allowed options: pool_id, aid_plate_reader). |
--readout-files |
ELISpot readout file(s). The expected columns are ‘plate_id’, ‘well_id’, and ‘spot_count’. If –readout-file-type is ‘aid_plate_reader’, then the readout-files are the Excel files from the AID plate reader machine. If –readout-file type is ‘aid_plate_reader’ and there were pools in 2 or more plates, then supply the files in the following order: plate 1 readout file, plate 2 readout file etc. |
--assignment-excel-file |
ELISpot assignment Excel file. Expected columns: ‘peptide_id’, ‘peptide_sequence’, ‘plate_id’, ‘well_id’ in a sheet named ‘block_assignment’. |
--min-pool-spot-count |
Minimum spot count for a pool to be considered a positive pool. |
--output-excel-file |
Output deconvolution Excel file. |
| Optional Parameter | Description |
|---|---|
--method |
Statistical deconvolution mode (default: ‘cem’). |
--background-spot-count |
Background spot count (default: ‘auto’). If ‘auto’, ACE automatically estimates the background spot count. The background spot count is used as negative control. Peptides with an estimated spot count less than or equal to the background spot count will be excluded as putative immunogenic hit peptides. Automatic estimation is applicable when the deconvolution method is constrained expectation-maximization (‘cem’). |
--verbose |
If True, prints messages. Otherwise, messages are not printed (default: True). |
Example
Let’s say you used the generate command in ACE to generate an ELISpot configuration for pooling 5 peptides per pool for a total of 25 unique peptides. The configuration also repeated each peptide 3 times (i.e. 3x coverage); you created 25peptides_5perpool_3x_configuration.xlsx. You took this configuration and successfully ran an ELISpot experiment. Now you want to deconvolve hit peptides. Here we describe how you can achieve this using ACE.
Any Reader
You can supply a generic ELISpot results .xlsx file to ACE:
25peptides_5perpool_3x_readout_pool-ids.xlsx:
| plate_id | well_id | spot_count |
|---|---|---|
| 1 | A1 | 0 |
| 1 | A2 | 300 |
| … | … | … |
| 1 | B3 | 0 |
Deconvolve hit peptides from your ELISpot experiment:
ace deconvolve \
--readout-file-type pool_id \
--readout-files ../test/data/25peptides_5perpool_3x_pool-id_readout.xlsx \
--assignment-excel-file ../test/data/25peptides_5perpool_3x_configuration.xlsx \
--min-positive-pool-spot-count 300 \
--output-excel-file outputs/25peptides_5perpool_3x_pool-id_readout_deconvolved.xlsxRunning the above command results in the following file:
AID Plate Reader
If you used a plate reader from AID, we are able to automatically parse the .xlsx file from the machine.
25peptides_5perpool_3x_readout_aid-plate-reader.xlsx:
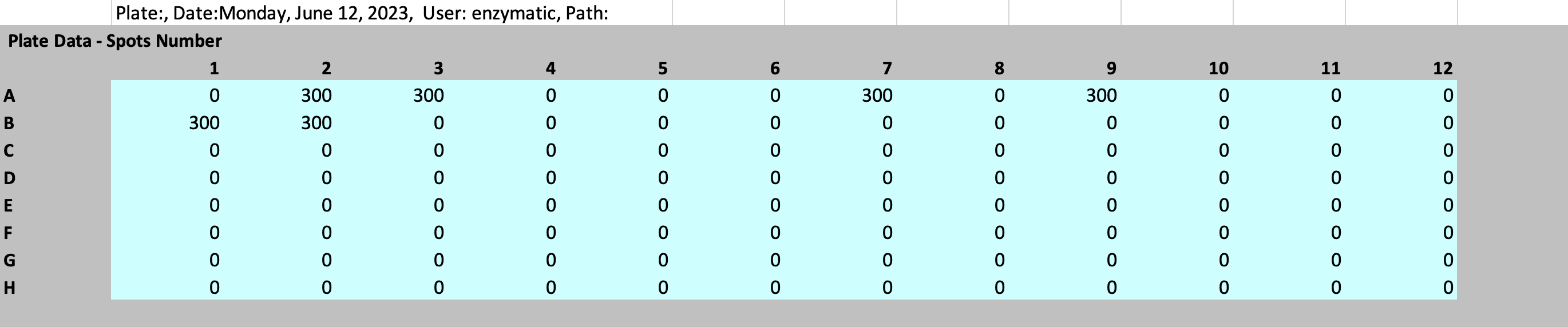
Deconvolve hit peptides from your ELISpot experiment:
ace deconvolve \
--readout-file-type aid_plate_reader \
--readout-files 25peptides_5perpool_3x_aid-plate-reader_readout.xlsx \
--assignment-excel-file ../test/data/25peptides_5perpool_3x_configuration.xlsx \
--min-positive-pool-spot-count 300 \
--output-excel-file 25peptides_5perpool_3x_aid-plate-reader_readout_deconvolved.xlsxRunning the above command results in the following file:
25peptides_5perpool_3x_readout_aid-plate-reader_deconvolved.xlsx:
Deconvolution results
ACE deconvolution identifies two classes of peptides: confident_hit and candidate_hit. Peptides that have the label confident_hit are confident hits because they were present in num_coverage (i.e. 3 in the above configuration) number of different pools with at least one unique assignment of hit peptide-pool. On the other hand, peptides that have the label candidate_hit need further validation (i.e. a second round of ELISpot) to determine whether it is indeed a hit peptide.